机器学习中的蛋白质折叠2025年3月17日 | 阅读 21 分钟  蛋白质就像我们体内的超级英雄,在支持我们组织、器官和全身过程的功能方面发挥着至关重要的作用。这些令人难以置信的分子由 20 种不同的构件组成,每一种都被称为氨基酸。令人难以置信的是,在我们体内存在着种类繁多的蛋白质,每种蛋白质都具有由数十甚至数百个氨基酸组成的独特序列。 最有趣的部分是,蛋白质中氨基酸的特定序列就像一个秘密代码,决定了它的超能力,例如它的功能。这个序列实际上决定了蛋白质的 3D 结构以及它在不同情况下的行为。你猜怎么着?这种独特的 3D 结构然后定义了蛋白质在各种生物过程中的特殊作用。所以,这不仅仅是一个普通的代码;它就像一个塑造蛋白质形式并释放其非凡功能的超级蓝图,使其成为我们身体运作方式的重要组成部分。 但这还不是全部!让我们深入探讨蛋白质折叠的迷人世界。想象一下:蛋白质就像艺术品,由长长的氨基酸链组成,它们的 3D 结构是释放其力量的关键。蛋白质折叠的过程就像一场复杂的舞蹈,蛋白质链优雅而精确地折叠成自己非凡且有功能的形状。这就像蛋白质发现了自己真正的身份,揭示了它独特的强大能力,以完成它在我们体内的使命。 然而,理解蛋白质折叠并非易事。鉴于该过程的巨大复杂性和蛋白质可以采用的无数种可能方式,这是一个难以解决的谜题。但科学家们正在追寻解开这个谜团,因为它对预测蛋白质结构至关重要,这对发现新药、研究疾病和进步生物工程等领域具有深远的影响。 理解它至关重要,因为它直接关系到蛋白质结构的预测。这种预测在药物发现、疾病研究和生物工程方面具有广泛的影响。然而,由于其复杂性和蛋白质可以采取的无数种构象,蛋白质折叠带来了挑战。 机器学习算法可以基于现有的蛋白质折叠数据进行训练,以学习蛋白质序列与其对应结构之间的模式和关系。然后,可以使用这些算法根据氨基酸序列预测新蛋白质的结构。通过分析已知蛋白质结构的庞大数据集,机器学习模型可以揭示控制蛋白质折叠的隐藏模式和原理。 机器学习在蛋白质折叠中的优势以下是机器学习在理解蛋白质折叠方面的一些优势
使用机器学习预测蛋白质折叠的缺点虽然使用机器学习预测蛋白质折叠具有许多优势,但这种方法也存在一些挑战和缺点,即
使用 Python 进行机器学习中的蛋白质折叠预测关于数据集该数据集包含从结构生物学协作研究中心 (RCSB) 蛋白质数据库 (PDB) 检索到的蛋白质信息。PDB 档案是一个庞大的数据集合,包括蛋白质和其他重要生物大分子的原子坐标和其他详细信息。为了确定分子中每个原子的位置,结构生物学家使用 X 射线晶体学、NMR 波谱和低温电子显微镜等各种方法。一旦获得这些信息,他们就会将其存入档案,由 wwPDB 进行注释并公开提供。 随着全球实验室研究的进展,PDB 档案不断发展。这使其成为研究人员和教育工作者的激动人心的资源。它提供了许多参与关键生命过程的蛋白质和核酸的结构,包括核糖体、癌基因、药物靶点,甚至整个病毒。然而,由于数据库的庞大,导航和查找特定信息可能会很困难。通常,一种分子有多种可用结构,或者结构是不完整的、经过修饰的,或者与其天然形式不同。 尽管存在挑战,PDB 档案仍然是科学界宝贵的数据来源,提供了有关各种生物分子结构的大量信息。研究人员和教育工作者可以探索这个庞大的存储库,以深入了解蛋白质和其他大分子的复杂性,支持结构生物学领域的进步。 内容有两个数据文件。两者都根据蛋白质的“structureId”进行排列
现在,我们将尝试构建一个模型来预测蛋白质结构。 代码
输出  我们利用 sidechainnet 来训练我们的机器学习模型,旨在根据给定的氨基酸序列预测蛋白质结构(角度或坐标)。这些示例几乎达到了全面模型训练所需的最低要求。 此处的代码默认设置为在调试数据集上进行训练。但是,您可以自由修改“scn.load”调用并选择其他 SidechainNet 数据集,例如 CASP12,进行进一步的实验和训练。 在这里,我们将使用两个简化的循环神经网络 (RNN) 来预测蛋白质的角度表示,使用它们对应的氨基酸序列。
内部 RNN 处理氨基酸序列,为每个氨基酸生成角度向量。虽然其他模型只使用了 3 个角度,但在我们的情况下,我们可以预测 SidechainNet 提供的所有 12 个角度。
请求 DataLoaders 时,您将收到一个字典,该字典将分割名称映射到相应的数据加载器。 输出  当批次被生成时,每个 DataLoader 会返回一个 Batch 名为 namedtuple 的对象,该对象具有以下属性:
输出  输出  输出 
辅助函数是代码中用于执行特定任务的小型、可重用代码片段。这些函数旨在简化复杂操作,提高代码的可读性,并避免代码重复。通过将复杂任务分解为更小、可管理的单元,辅助函数使主代码更加有序且易于维护。
注意力层在深度学习模型中很重要,因为它们有助于模型专注于数据中最相关部分。它们就像人类的注意力一样,在学习过程中,某些事物比其他事物更重要。
在这里,我们将训练模型,例如将二级蛋白质结构矩阵作为输入。 模型输入
PSSMPSSM,也称为位置特定评分矩阵或 DNA 上下文中的位置权重矩阵,表示一个矩阵,为序列中的每个位置提供特定的分数或概率。 这就像一个特殊的代码,告诉我们每个字母(氨基酸)出现在秘密消息(蛋白质序列)的不同位置的可能性。科学家们通过比较来自不同生物的许多相似秘密消息来创建这个代码。PSSM 帮助他们了解哪些字母很重要,哪些字母可以更改而不影响消息的含义。这就像拥有一个秘密解码器,可以帮助科学家们更多地了解蛋白质中的秘密消息以及它们的工作原理。 由于 PSSM 和序列都有 20 种不同的信息,二级结构有 8 种可能性,信息量是每个部分的单个数字;当我们将所有这些放在一起时,我们需要总共 49 个值来正确表示它们。 输出  输出   输出  输出 
在许多情况下,我们使用scn.BatchedStructureBuilder,它需要两项内容:
我们有一个知道如何猜测某些角度的正弦和余弦值的模型。但我们需要实际的角度,而不是正弦和余弦值。因此,我们使用一个名为scn.structure.inverse_trig_transform的特殊工具将正弦和余弦值转换回实际角度。一旦我们有了实际角度,就可以将它们提供给 BatchedStructureBuilder。
在这里,我们将模型的预测蛋白质结构与实际蛋白质结构进行比较。为了便于理解,我们使用 3D 图进行可视化比较。每个示例都有两个图:上面的图显示了模型对蛋白质结构的预测,下面的图显示了实际的蛋白质结构。这使我们能够看到模型的预测与实际蛋白质结构匹配的程度。 示例 (01) 输出 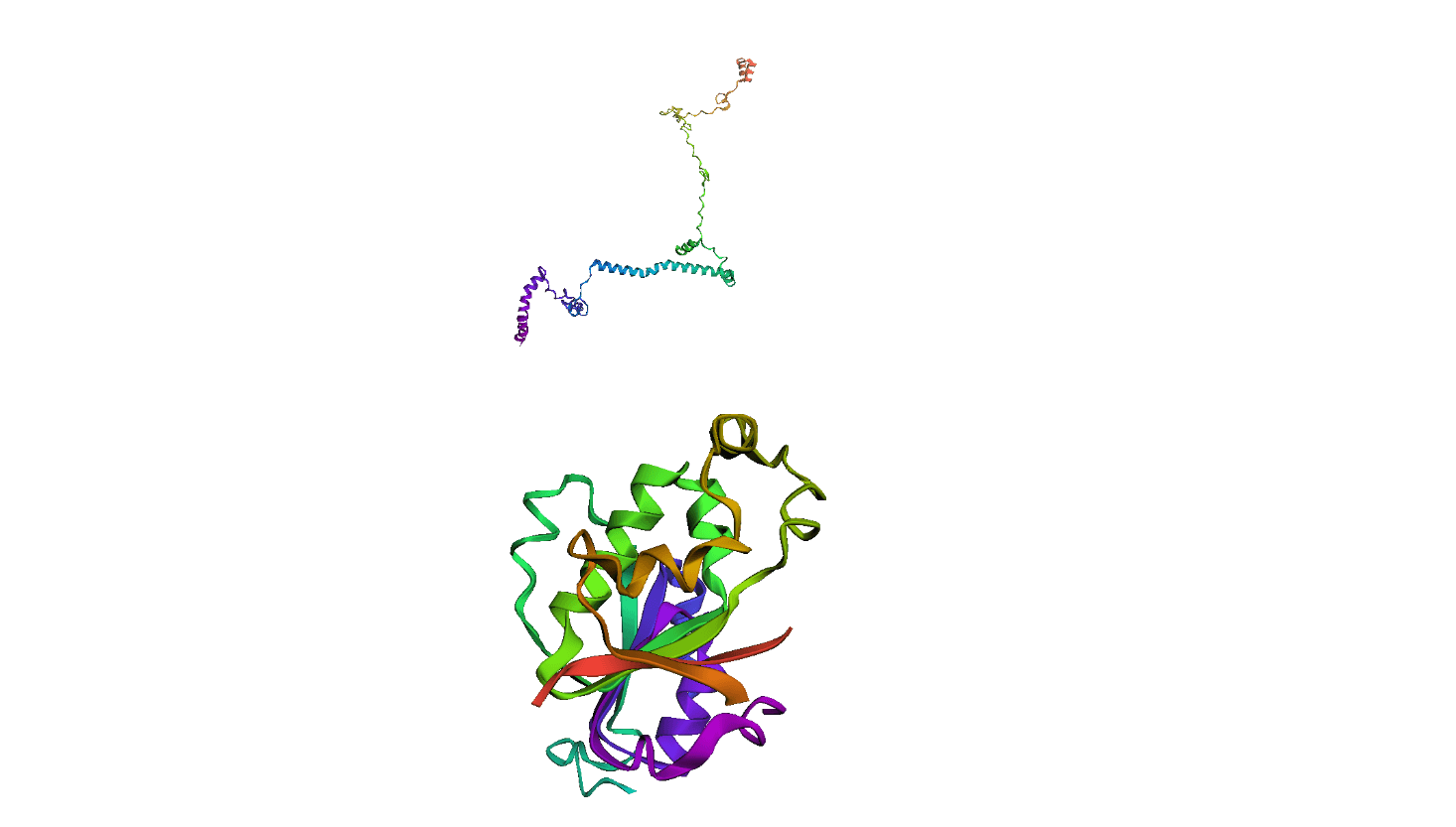 示例 (02) 输出  示例 (03) 输出  示例 (04) 输出  示例 (05) 输出  示例 (06) 输出  输出  输出  训练 (序列 → 角度)现在我们将训练模型,同时将蛋白质序列作为输入。 信息流:这里的信息流在一个简单的 Transformer(Attention)模型中,该模型处理序列数据。输入表示为 [Layers*21],经过 Embedding 层,得到 [Layers. Dense Embedding]。然后,它通过 LSTM 层,转换为 [Layers. Dense Hidden]。最后,输出从 LSTM 出来并通过 [Layers Dense Output] 层。在此过程中,模型处理输入数据,提取相关信息并生成最终输出,而不会对其进行修改。 处理角度的圆形性质:为了帮助我们的模型理解角度 π 和 -π 是相同的,我们使用了一个特殊的技巧。我们不直接预测角度,而是为每个角度预测两个值:sin 和 cos。然后,我们使用 atan2 函数将这两个值组合起来恢复角度。这样,模型的输出形状将为 L×12×2,其中 L 是蛋白质序列的长度,值为 -1 到 1 之间。这种方法使我们能够正确处理角度并提高预测的准确性。 输出   输出  输出  推理 (序列 → 角度)示例 (09) 输出  示例 (10) 输出  示例 (11) 输出  示例 (12) 输出 
使用机器学习进行蛋白质折叠的未来展望机器学习在蛋白质折叠领域的未来潜力令人难以置信,它有能力改变我们对蛋白质结构和功能的理解。通过利用机器学习的能力并采取跨学科战略,我们正站在发现新机遇和拓展科学探索边界的门槛上。当我们继续解开围绕蛋白质折叠的谜团时,我们踏上了一条革命性研究和开创性应用的道路,这将对人类福祉及其他方面产生深远影响。 结论蛋白质折叠是一个关键而复杂的过程,它深刻地影响着蛋白质的行为和功能。机器学习与生物信息学的融合提供了一条令人兴奋的途径来深入研究这个复杂的世界,使我们能够以前所未有的精度预测蛋白质结构。机器学习和生物信息学之旅有望带来变革性的发现,将彻底改变医学和生物技术。当我们向前迈进时,蛋白质折叠的谜团将逐渐被解开,揭示生命本身的深刻复杂性。有了机器学习作为我们的盟友,我们离揭开蛋白质折叠的秘密及其在生命宏大织锦中的广泛含义越来越近了。 下一个主题使用机器学习进行情感分析 |
我们请求您订阅我们的新闻通讯以获取最新更新。
我们提供所有技术(如 Java 教程、Android、Java 框架)的教程和面试问题
G-13, 2nd Floor, Sec-3, Noida, UP, 201301, India
